docker-overview
Overview of the Docker container system

Sheffield R meetup - 6th March 2018 (Updated for SITraN journal club - 2nd April 2019)
Longer version of materials prepared for CRUK Cambridge available here
Mark Dunning (@DrMarkDunning) Bioinformatics Core Director
Sheffield Bioinformatics Core
web : sbc.shef.ac.uk
twitter: @SheffBioinfCore
email: bioinformatics-core@sheffield.ac.uk
Basics
https://docs.docker.com/engine/docker-overview
Docker is an open platform for developers to build and ship applications, whether on laptops, servers in a data center, or the cloud.
- Or, it is a (relatively) painless way for you to install and try out Bioinformatics software.
- You can think of it as an isolated environment inside your exising operating system where you can install and run software without messing with the main OS
- potentially a good way for beginners to learn command-line tools?
- Really useful for testing software
- Clear benefits for working reproducibly
- instead of just distributing the code used for a paper, you can effectively share the computer you did the analysis on
- For those of you that have used Virtual Machines, it is a similar concept
- However, they are more lightweight and easier to distribute
- Images are combined in a layered system
Installing Docker
Mac
Windows
(may require some messing around with virtualisation or Hyper-V)
Once you have installed Docker using the insructions above, you can open a terminal (Mac) or command prompt (Windows) and run the following to download an image for the Ubuntu operating system from Dockerhub;
docker pull ubuntu
N.B. on Linux, you may need to run docker with sudo, unless you apply this fix
To run a command inside this new environment software we can do;
docker run ubuntu echo "Hello World"
![]()
![]()
- run the docker container for the Ubuntu operating system
- run the
echocommand within this operating system - exit
To use the container in interactive mode we have to specify a -it argument. Which basically means that it doesn’t exit straight away, but instead runs the bash command to get a terminal prompt
docker run -it --rm ubuntu
- the
--rmmeans that the container is deleted on exit, otherwise your disk could get clogged up with lots of exited containers -
if no command is specified, you get a shell prompt
- the
ubuntuimage (orcentos) is often used as a base image upon which other more complicated images are based - when you want another image, you only have to download the changes that have been made
+ i.e. don’t need to download
ubuntuagain - more compact images, easier to distribute
- compare to virtual machine
Volumes in Docker
You’ll notice that when you launch a container, you don’t automatically have access to the files on your OS. In Docker, we can mount volumes using the -v argument to make files accessible e.g. -v /PATH/TO/YOUR/data:/data inside the container.
## should say that no file or directory exists
docker run --rm ubuntu ls /data
## If on Windows, need correct path separator
docker run --rm -v c:\work:/data ubuntu ls /data
## On Unix it would be something more sensible, like
docker run --rm -v c/home/USER/work:/data ubuntu ls /data
Running R (and RStudio) through Docker
The latest version of R and R devel are provided by the rocker project https://github.com/rocker-org/rocker
docker run --rm -it r-base R
- pull the latest
r-baseimage, if you don’t have it - run interatively (
-it) - run the
r-basedocker image - run the
Rexecutable
For latest developmental version of R:-
docker run --rm -it r-devel R
Can also get previous versions of R
- good if you need to re-run code that was written on a previous R version
- good if you need to test code on latest version of R
RStudio is also supported. See https://github.com/rocker-org/rocker/wiki/Using-the-RStudio-image
- this time we do something slightly different
docker run -p 8787:8787 rocker/rstudio
- the
-pargument opens a port in the docker container - open a web browser and enter the address
http://localhost:8787 - username
rstudiopasswordrstudio - you have a version of RStudio working in your web browser!
You can install whatever R packages you need in this container and analyse your data
N.B. Python fans needn’t feel left out; there are docker containers for jupyter too.
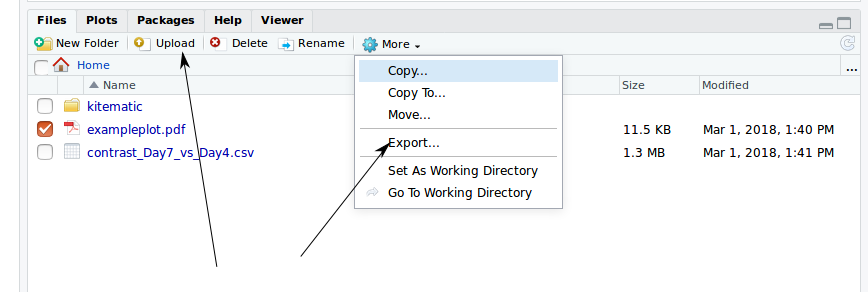
- you can mount a volume with
-v.- or there is an Upload option in the file viewer
- data (and scripts etc) can be exported with the Export menu item
Once a docker container has quit, you can jump back in with docker start and docker attach
docker ps ##not name of container that just quit
docker start <name-of-container-that-just-exited>
docker attach <name-of-container-that-just-exited>
You can then build a new image
docker commit <name-of-container-that-just-exited> <new image>
There may already be a docker container for popular sets of tools
- e.g. the tidyverse
- bioconductor
docker run --rm -p 8787:8787 -e PASSWORD=PASSWORD bioconductor/release_core2
The Dockerfile
The creation of Docker images is specified by a Dockerfile. This is a text file containing the sequence of instructions required to re-create your image from some starting point, which could be the standard Ubuntu image. Essentially we list the commands we use, step-by-step to install all the software required. If you already have a shell script to install your software, then translating to a Dockerfile is relatively painless.
A useful reference is the official Docker documentation on Dockerfiles, which goes into far more detail than we will here.
The example below shows the Dockerfile used to create a Ubuntu image use git to clone a repository and install some packages
FROM ubuntu
MAINTAINER YOU NAME<your.name@sheffield.ac.uk>
RUN apt-get update
RUN apt-get install -y wget build-essential git
RUN git clone.....
RUN R -e 'install.packages(....)'
The docker build command will build a new image from a Dockerfile. With docker push you can distribute this on dockerhub once you have a user name.
docker build -t=my_username/my_new_image .
docker push
Use Case 1:- Distributing software for a training course
Several headaches can emerge when preparing the materials for a training course
- if the course venue has no desktop machines, participants will need to bring their own machines
- so they will need to install software beforehand
- challenging for beginners
- docker presents a potential solution
- (however, they will still need to install docker - which could still be a barrier for some)
- distributing materials to other participants who can’t make the class, or who want to attend remotely
- when developing materials in a team, need to agree on common software versions etc
- could pre-install the container on an AWS instance
docker pull markdunning/rnaseq-toolkit docker run --rm -p 6080:80 markdunning/rnaseq-toolkit
Use Case 2:- Distributing supplementary data for a publication
- Stephen Eglen of Department of Applied Mathematics and Theoretical Physics, University of Cambridge made the data and code for his paper available on github

- furthermore, the scripts, data are available with the appropriate version of R as a docker container
docker run -d -p 8787:8787 sje30/waverepo
Use case 3:- Reproducible analysis
Issue: doing several analyses at same time, some of which may require latest version of R etc. How can we ensure that previous analyses still run. Within each project, create a Dockerfile to build a container for the analysis.
FROM bioconductor/release_base2:R3.5.3_Bioc3.8
MAINTAINER Mark Dunning<m.j.dunning@sheffield.ac.uk>
RUN R -e 'install.packages("BiocManager")'
RUN R -e 'BiocManager::install("tidyverse")'
RUN R -e 'BiocManager::install("DEXSeq")'
Useful docker commands
To see what containers you have run recently
docker ps -a
If you find your disk filling up with docker images, there are convenient one-liners for removing all containers and images.
Don’t run this now, unless you want everything you’ve been working on to be deleted!
docker rm $(docker ps -a -q)
docker rmi $(docker images -q)
You can go back into the environment of a container that has been exited. Firstly, we make sure the container is running by using docker start:-
docker start <container_ID>
We can then use docker attach. Note that you will have to press ENTER twice in order to get a new command prompt within the container.
docker attach <container_ID>
The elephant in the room…
Sounds great so far! But…
- when you run a Docker container you have super-user access rights inside that container. Unix admin people that manage HPC systems don’t like this.
There is an alternative….
Singularity
![]()
You can build a singularity image from a docker container
- Use the docker image on dockerhub at
markdunning/dexseq-analysis(build fromDockerfileabove) to build an image. You need sudo access to do this. - Copy the image to
sharc - Run the
execcommand to RunRwith an analysis script
### Run where you have sudo access
sudo singularity build singularity/dexseq docker://markdunning/dexseq-analysis
## Copy the image to sharc /shared/bioinformatics_core1/Shared/software/singularity/dexseq
## On sharc
singularity exec /shared/bioinformatics_core1/Shared/software/singularity/dexseq R -f dexseq_analysis.R
More documentation is available for using singularity on sharc